
I've been using phred/phrap/consed for a few months now. I am now encountering a strange failure of the package to do an assembly/alignment
In my (mini)assembly, I have three contigs (100Kbp, 75Kbp, 900bp) as well as four sanger sequencing reads (from ab1 files). Consed is matching a 152bp region between the two largest somewhat near the ends (55bp from the end of the 75Kb contig, 1766bp from the end of the 100Kb contig) - a region that only has 2 mismatches between the two contigs. It seems somewhat rational to join there, but in no prior assemblies or miniassemblies did it attempt to make this join (pending verification I believe them to be separated by just under 4KB). However, the 900bp contig is being placed as only aligned to 10 consecutive bases of the 75Kb contig, but is embedded therein (near the junction with the largest contig). Three of the Sanger reads are "aligned" to the 75Kbp contig were I don't see ANY significant congruence between the reads and the contig. Further, one of these three reads is offset 16bp from the other two (screenshot). The fourth sanger read is "overlapping" in part of the 2Kb mismatch region between the two largest contigs, but shows no sequence similarity to either at this location. Additionally, no gaps exist in any of the sequences or in the "consensus" in the assembly. In short, sequences are not aligning, nor are gaps being inserted in reads to establish an alignment, yet all are being assembled into a single contig.
I've used the miniassembly option many times in the past without issue, one thing that changed was I've recently also started using assembly view to merge contigs semi-manually. However, these contigs that I'm attempting to join are unadulterated by the assembly view.
The phrap command-line the miniassembly runs is, by default,
/usr/bin/genome/bin/phrap.longreads mini.120907.103603.fasta.screen -new_ace -view -retain_duplicates -trim_qual 14 -trim_start 0 -repeat_stringency .95 -forcelevel 0 -bypasslevel 0 -maxgap 30 -minmatch 14 -minscore 35 -maxmatch 40 -vector_bound 30 -max_subclone_size 8000
During crossmatch I get this error: NO QUALITY FILE blahOld.contigs.qual WAS FOUND. REMAINING INPUT QUALITIES SET TO 15. Done
despite all contigs having associated quality values in the screen.ace file and all Sanger sequences converted by p/p/c from .ab1 to both phd and scf.
In my (mini)assembly, I have three contigs (100Kbp, 75Kbp, 900bp) as well as four sanger sequencing reads (from ab1 files). Consed is matching a 152bp region between the two largest somewhat near the ends (55bp from the end of the 75Kb contig, 1766bp from the end of the 100Kb contig) - a region that only has 2 mismatches between the two contigs. It seems somewhat rational to join there, but in no prior assemblies or miniassemblies did it attempt to make this join (pending verification I believe them to be separated by just under 4KB). However, the 900bp contig is being placed as only aligned to 10 consecutive bases of the 75Kb contig, but is embedded therein (near the junction with the largest contig). Three of the Sanger reads are "aligned" to the 75Kbp contig were I don't see ANY significant congruence between the reads and the contig. Further, one of these three reads is offset 16bp from the other two (screenshot). The fourth sanger read is "overlapping" in part of the 2Kb mismatch region between the two largest contigs, but shows no sequence similarity to either at this location. Additionally, no gaps exist in any of the sequences or in the "consensus" in the assembly. In short, sequences are not aligning, nor are gaps being inserted in reads to establish an alignment, yet all are being assembled into a single contig.
I've used the miniassembly option many times in the past without issue, one thing that changed was I've recently also started using assembly view to merge contigs semi-manually. However, these contigs that I'm attempting to join are unadulterated by the assembly view.
The phrap command-line the miniassembly runs is, by default,
/usr/bin/genome/bin/phrap.longreads mini.120907.103603.fasta.screen -new_ace -view -retain_duplicates -trim_qual 14 -trim_start 0 -repeat_stringency .95 -forcelevel 0 -bypasslevel 0 -maxgap 30 -minmatch 14 -minscore 35 -maxmatch 40 -vector_bound 30 -max_subclone_size 8000
During crossmatch I get this error: NO QUALITY FILE blahOld.contigs.qual WAS FOUND. REMAINING INPUT QUALITIES SET TO 15. Done
despite all contigs having associated quality values in the screen.ace file and all Sanger sequences converted by p/p/c from .ab1 to both phd and scf.

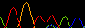

Comment