Originally posted by dnusol
View Post
-
Maybe RNAseq is biased by nature?
Check these other threads
Leave a comment:
-
Hi, rhinoceros, here are more details about the samples.Originally posted by rhinoceros View Post
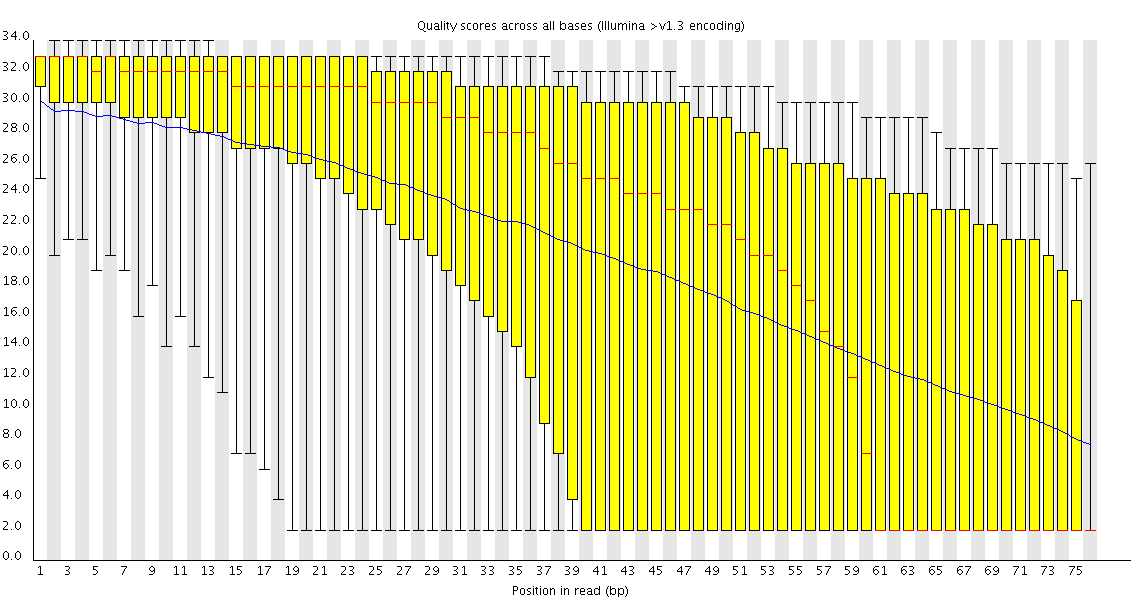
The total RNA was extracted from one piece of mouse muscle by Qiagen RNeasy fibrous tissue mini kit. The quality of RNA were estimated by Agilent bioanalyzer. A260/A280 for all samples were around 2.0. RIN for all RNAs were around 10. It seems the quality of RNAs is excellent. And I didn't seem a chance to have other chromosomes.
The Libraries were prepared by a RNAseq center with an illumina TruSeq kit, but I know little about that...
Here is the whole FastQC report, which may give you more information. http://zgan.weebly.com/uploads/8/9/3...tqc_report.zip
More clues for me? ThanksLast edited by ZoeG; 06-18-2013, 03:53 PM.Leave a comment:
-
Thanks, dnusol. I went through the webpage, what I can tell is that the library seems something wrong. The quality score actually seems okay. When I mapped the data to its reference, >90% reads were mapped successfully. Do this result conflict with the conclusion of wrong library? Confused..Originally posted by dnusol View PostLeave a comment:
-
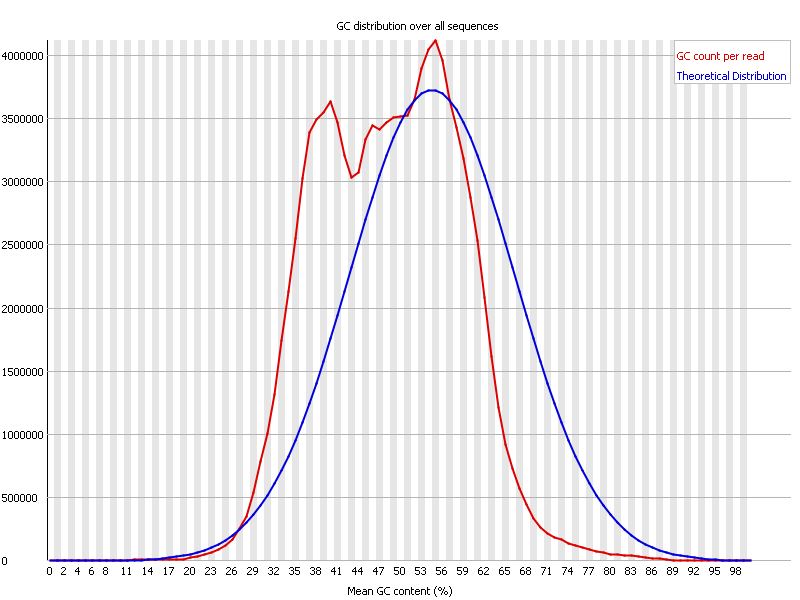
Looks to me like the reads come from two very different sources (the two peaks). Perhaps even three sources.. I don't know. How are we supposed to tell since you didn't specify what was sequenced. Since the read counts (of the peaks) are somewhat similar, perhaps your source has two chromosomes, which have different GC (not unheard of).Last edited by rhinoceros; 06-14-2013, 04:23 AM.Leave a comment:
-
This link might be useful
Leave a comment:
-
Contamination? What does this per sequence GC content mean?
Hello, I am quite new to RNAseq analysis.
When I did the quality control for my Illumina HiSeq data, I found an abnormal per sequence GC content, as the picture shows.
It seems some contamination?
Need help, thanks
-
The complexity of cancer is clearly demonstrated in the diverse ecosystem of the tumor microenvironment (TME). The TME is made up of numerous cell types and its development begins with the changes that happen during oncogenesis. “Genomic mutations, copy number changes, epigenetic alterations, and alternative gene expression occur to varying degrees within the affected tumor cells,” explained Andrea O’Hara, Ph.D., Strategic Technical Specialist at Azenta. “As...07-08-2024, 03:19 PM
Leave a comment: