 Tweet
Tweet
Originally posted by Yilong Li
View Post
-
The next release of the program will feature some improvements to try to make the graphs easier to interpret. I'll add in horizontal lines from the y-axis points as well as I can see that this would make life easier. -
FastQC v0.8.0 has been released. This features improved graphs (from some of the suggestions presented here). It also adds an option to analyse only mapped entries from BAM/SAM files which should make life easier for colorspace people, as well as adding an option to run multiple threads to process files in parallel.
You can get the new version from:
[If you don't see the new version of any page hit shift+refresh to force our cache to update]Comment
-
quality control for pair-end analysis
Dear Simon,
I am also using your tool, which is great by the way. I am starting with my first pair-end experiment from illumina. I am wondering if I should look at each fastq file generated for each lane (_1 and _2) separately or I should merge them into one single file and run the script. They come from the same lane so I suspect you would want to have them merged to get one QC value for each lane, right?
Thanks,
DaveComment
-
We analyse our first and second read data separately. Although they come from the same insert there could easily be a problem with affected only the first or second read, and which would be difficult to spot if you concatonated the two files.Originally posted by dnusol View Post
If you did want to combine then it would be better to conactonate the individual reads together so read 2 followed on from read 1 in the same sequence so you wouldn't lose any information, but I'm not sure that's worth the hassle.
This can actually be a problem with BAM/SAM files containing paired end data where we currently can't separate out systematic biases which affect only one of the reads. I did look at adding an option to separate these into separate reports but didn't come up with a simple way to do this which didn't feel clunky.Comment
-
Dear Simon,
in this link
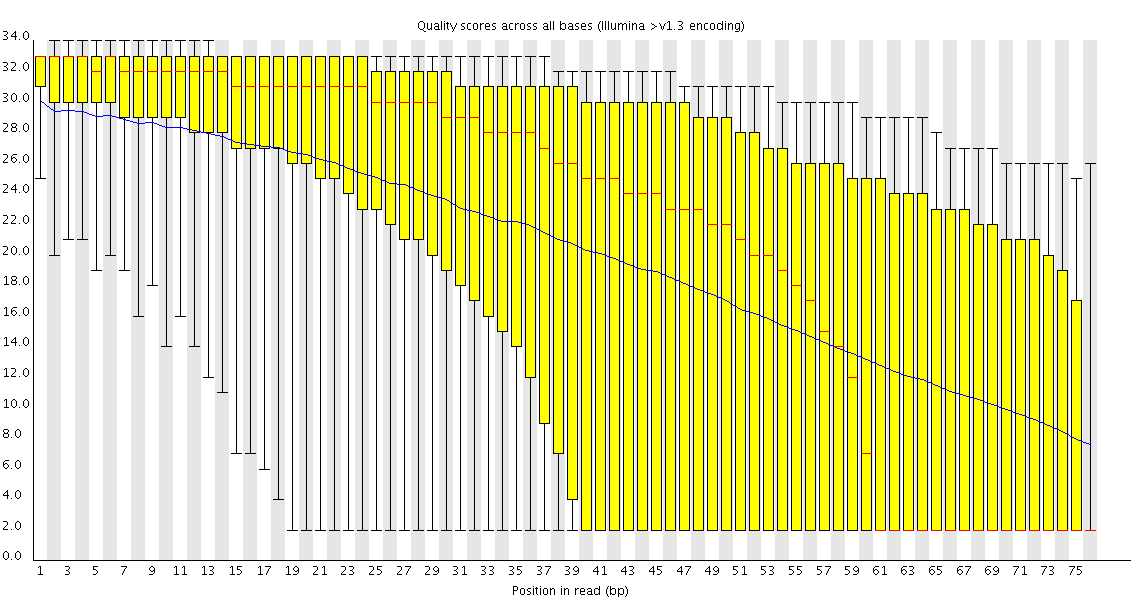
you showed an image of a run with quality dropping gradually. You suggest trimming. I am having the same problem with the paired-end experiment I mentioned before and instead of trimming I thought of filtering reads that did not pass a quality threshold. But this filter would probably have to work on both s_N_1_sequence.txt and s_N_2_sequence.txt simultaneously taking into account the pairing of the reads. Do you know of any tool that could do this? Or is it better to trim both files similarly to a desired length so that the pairing is not lost?
Thanks,
DaveComment
-
Whether and how you trim poor quality sequence is going to depend both on how poor your quality gets, and what you're going to do with the data. In general I don't filter poor quality reads unless I have a specific reason to. Many downstream applications can take the quality into account when they work so will effectively ignore the poor data for you. For some specific applications (bisulphite sequencing is the obvious example, but some SNP calling applications would be similar), removing poor quality data makes your life much easier as false calls can have a disastrous effect if left in in any quantity - but these are the exception rather than the rule.
In the run I linked to the quality dropped to such an extent that the bases much above 50 were effectively useless so I wouldn't worry about losing useful information by removing them. If my run had only been 55 bases I might well have kept everything.
Removing reads based on their average quality can be useful where you see a specific subset of poor quality data (FastQC will show you this in the per-read quality plot), but often you will see a general loss of quality over the length of the read so you'd be throwing away good data in many cases. I'm not aware of a tool which would filter on paired reads quality but some of the toolkits could probably be easily adapted to do it.
You normally find that poor quality in one read will be reflected in its pair, but there's no intrinsic problem with having paired ends with different lengths if you had a problem at only one end (unless your downstream application doesn't handle this well).Comment
-
FastQC v0.9.0 has been released. The major change is that this version now has much better support for the analysis of datasets containing long and variable read lengths.
Many of the plots which were ususable before on this kind of data are now grouped into variable sized bins so the graphs can effectively summarise even the longest and most diverse datasets. This will primarily affect users of long (>75bp) Illumina reads but especially 454 or PacBio data.
To see what the changes look like I've put up an example report from a 454 and PacBio dataset on the fastqc project page.
You can get the new version from:
[If you don't see the new version of any page hit shift+refresh to force our cache to update]Comment
-
Hi Simon and others,
I have a question regarding thresholds for the per sequence quality scores.
We did a lot of whole exomes on our Illumina machines and for each sample I generate a QC report. I have incorporated some of the FastQC outcome as well, including the number of reads (as a percentage of the total number of reads) that have a mean quality of <27 (I sum the counts for score < 27 from the per sequence quality scores module in the fastqc_data.txt file). I used 27 as a threshold because it's also used by the per sequence quality scores module.
But Simon why did you choose 27? What would be a reasonable error rate threshold to report in my situation? What do you and others say?
Cheers,
Bruins
(and, naturally, thanks again for the awesome tool!!)Comment
-
The thresholds used in FastQC are somewhat arbitrary and are simply based on looking at a load of data coming out of our sequencing facility and public datasets. I've generally picked thresholds which equate to the points where I'd have been concerned if I saw data with those properties. They've mostly been based on Illumina data, but more recently I've run a lot of data from other platforms through and the results seemed mostly sensible there as well.Originally posted by Bruins View Post
I suspect that for something like Phred scores there isn't going to be a single ideal answer for where to set a cutoff which would apply to all run variants on all platforms. Data which might appear to be of poor quality if it came from a short Illumina run would be fantastically good if it came from a PacBio for example.
I've tried to stress that the pass/warn/fail levels in FastQC are not absolute limits but are intended to be indicative of aspects of your data which you might want to investigate further (or at least be aware of) rather than a flag to say that if you see this you should throw it away.
To address your specific question - I've actually been thinking about the per-sequence quality plot lately. This is actually the plot where I'm least happy about the metrics used to evaluate the pass/warn/fail levels. At the moment we just find the peak in this graph and see where it lies, but I've seen too many datasets where this simple measure fails to spot what seem to be obvious problems in the data. In particular I would expect to see a particular profile of mean quality scores (looking like a skewed normal distribution), and I'd like to spot deviations from this distribution so that secondary peaks with lower quality would trigger a warning. Finding the right way to model and measure this is something I'm hoping to find some time to look at.
I'm *very* keen to hear feedback about both the metrics and the cutoffs used in the program and have adjusted things in response to previous suggestions. However it's often difficult to find a good balance where you have reasonably stringent quality criteria but you don't end up giving everyone the impression that their data is rubbish.Comment
-
Hello
i'm new here, and i started to try to use this program, but i get an error: you can see here: http://img27.imageshack.us/f/semttulohl.png/
it says that my ID line didn't start with "@", i can change the original file and put an "@" in the beggining?Comment
-
It looks like you're trying to use a file which isn't in FastQ format. I'd guess from the name of your file that it's a fastA file? FastQC is designed to work with file formats which include both sequence and quality data, which fastA doesn't have. You could, I suppose, make up a fake FastQ format from your fastA file if you really wanted to, but it would probably be better to find your raw sequence output and run that through the program, rather than trying to analyse your assembled contigs directly.Originally posted by Chuckytah View Post
If you let us know what kind of data you have and what you're trying to find out we may be able to offer other suggestions.Comment
-
Hi,
what kind of formate are the sequences you are working with?
the "@" sign means the seq. suppose to be in a fastq formate.
and not in the fastA formate which begins with a ">".
There are scripts to convert from one to the other. Like this one:
http://seqanswers.com/forums/showthr...ht=fasta2fastq
but you'll also need a quality file of your data.Comment
-
Originally posted by frymor View Post
Those scripts i need to instal C and/or Perl to execute them, isn't there any program that convert it instantaneosly? without needing to instal the languages?
sorry my dumbness, but i'm new to bioinformatics field... :SComment
-
What's the file format you are using ? Looks like there's no header within your sam file. Have you tried it with a fragment of your file or just send the first 10 lines of your file.
Cheers
MComment
-
I have .txt and .xlx files... but i'm using the .txt cause it's in FASTA format.Originally posted by Martin R View PostComment

-
The field of immunogenetics explores how genetic variations influence immune responses and susceptibility to disease. In a recent SEQanswers webinar, Oscar Rodriguez, Ph.D., Postdoctoral Researcher at the University of Louisville, and Ruben Martínez Barricarte, Ph.D., Assistant Professor of Medicine at Vanderbilt University, shared recent advancements in immunogenetics. This article discusses their research on genetic variation in antibody loci, antibody production processes,...11-06-2024, 07:24 PM -
Next-generation sequencing (NGS) and quantitative polymerase chain reaction (qPCR) are essential techniques for investigating the genome, transcriptome, and epigenome. In many cases, choosing the appropriate technique is straightforward, but in others, it can be more challenging to determine the most effective option. A simple distinction is that smaller, more focused projects are typically better suited for qPCR, while larger, more complex datasets benefit from NGS. However,...10-18-2024, 07:11 AM
Comment