
 Tweet
Tweet
Originally posted by JVGen
View Post
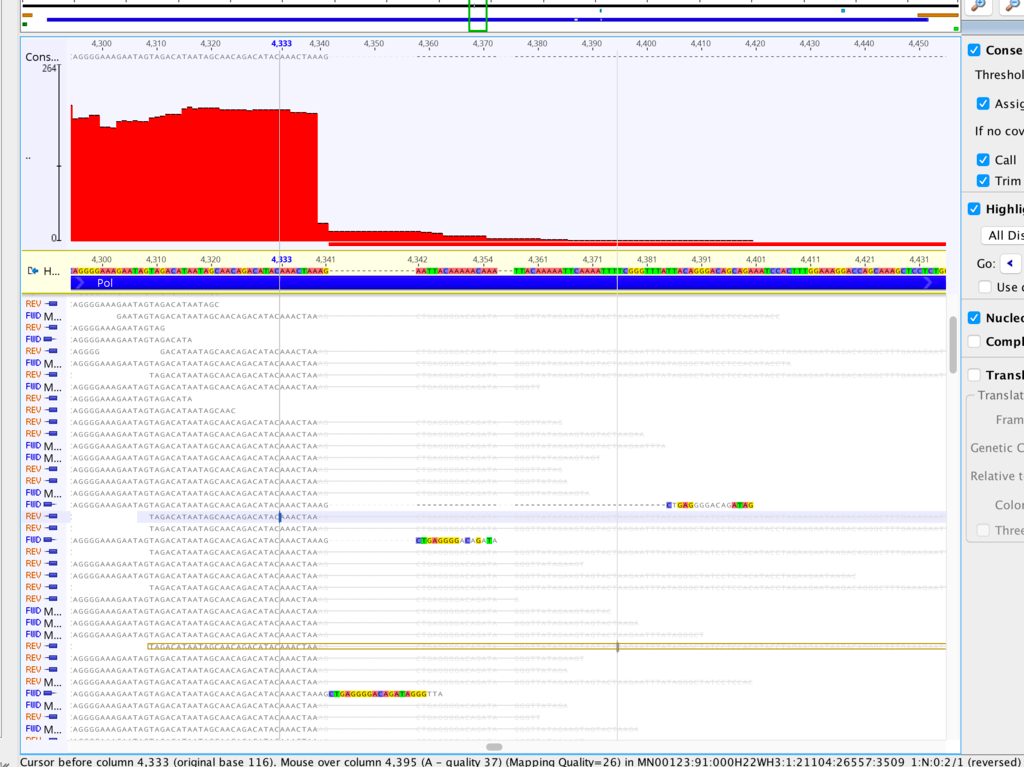
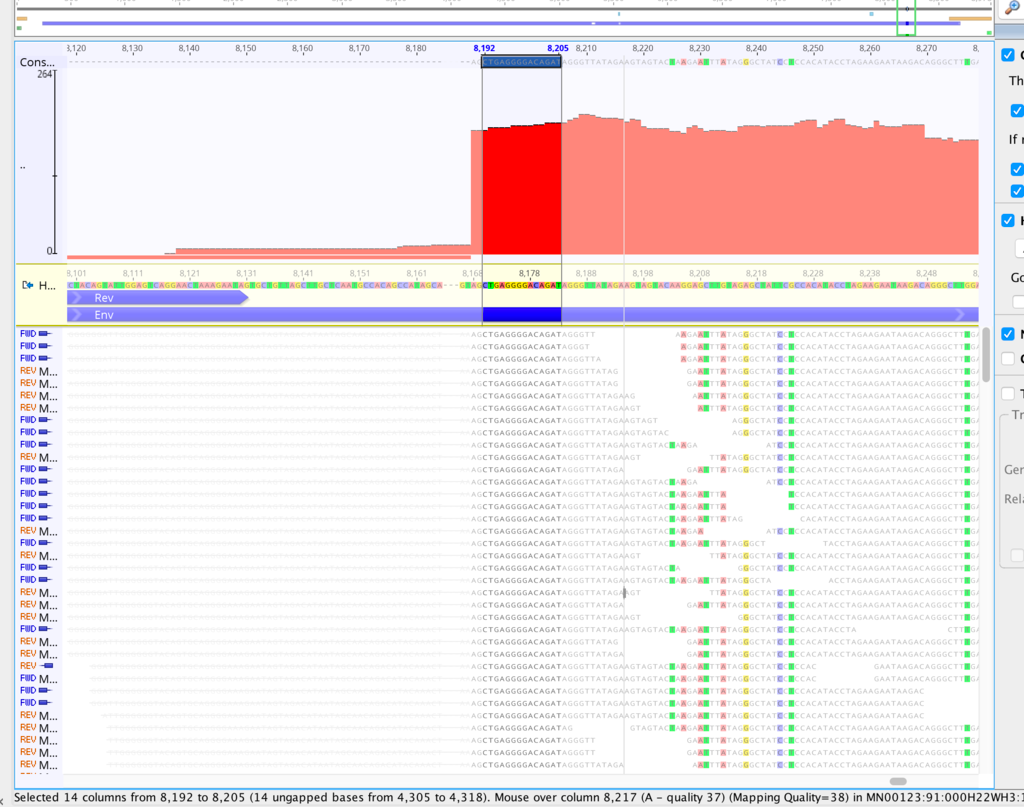
It might be helpful in this case if you could generate a kmer frequency histogram to see whether the contaminant and non-contaminant sequence is easily separable by depth alone. If so, there are a couple of easy ways to remove it. You can generate a kmer-frequency histogram with kmercountexact or BBNorm; just attach the text file to this thread. Normally I look at it in a log-log plot.
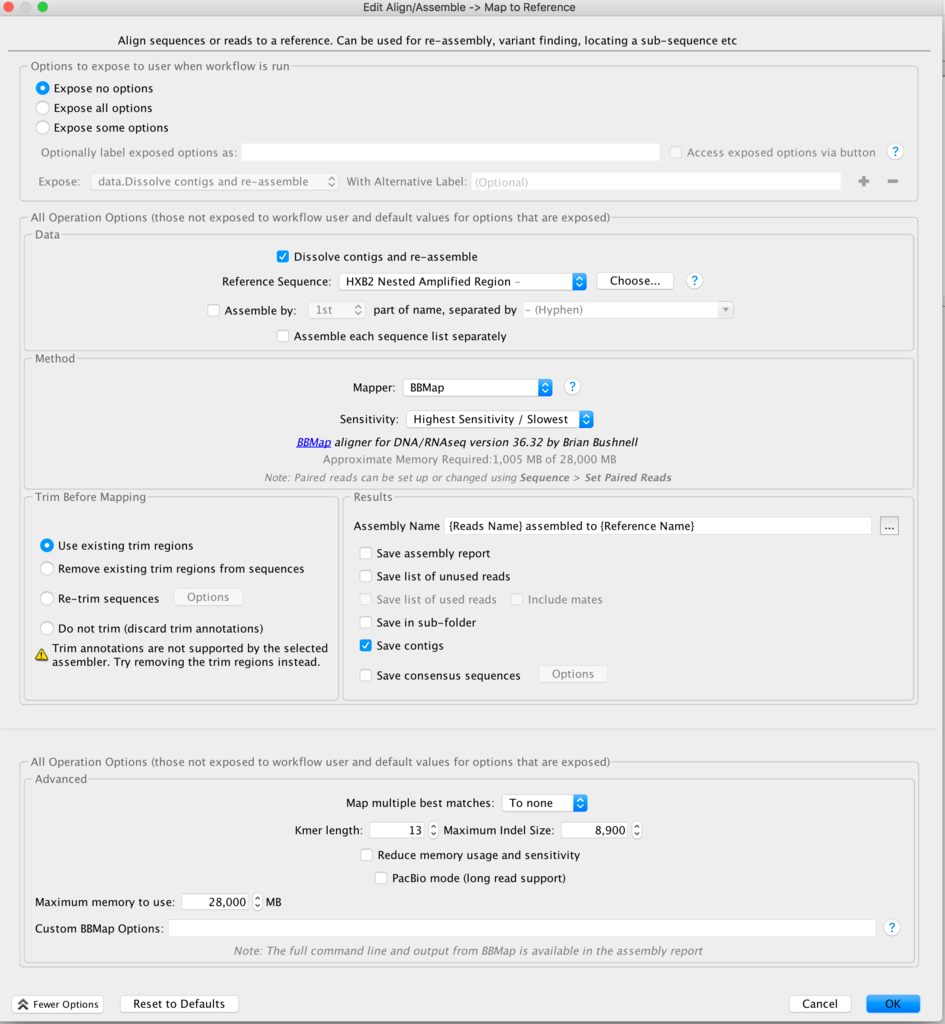
What assembler are you currently using, by the way? I've had poor results with Spades on viruses, and better results with Tadpole. But this was raw viral sequence and amplicon sequence may give different results.
As for consultancy, I've sent you a pm.
-Brian





Comment