
 Tweet
Tweet
Originally posted by lh3
View Post
I beg to disagree with you on one point: of all the *-seqs, RNA-seq is probably the one that cares about the CIGAR string the most. Splice junctions provide crucial information about expression of isoforms (alternative splicing). Unlike indels, splice junctions are very abundant in long RNA libraries, and with longer reads we are seeing >50% of read pairs with at least one splice, so realignment would not be practical. Also, standard global alignment algorithms will not be feasible, I believe, since junctions span tens of kilobases in mammalian genomes.
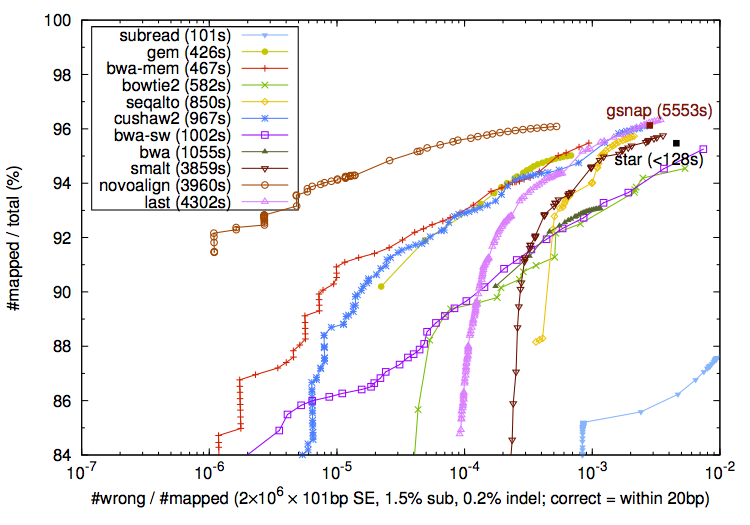
On the other hand I agree that aligners should not be judged harshly for soft-clipping a few bases from the ends of the reads when these bases cannot be placed confidently. In addition to the standard "correctly mapped locus" metric, one could imagine a metric which would count all correctly (true positive) and incorrectly (false positive) mapped bases for each read - that would show which aligners perform better for recovering the entire alignment.
Cheers
Alex






Comment