-
i use bowtie2 and the result show that the uniq-map is " 24824681 (15.15%) aligned concordantly exactly 1 time",i think it is too small and i will try to use bwa.My reference is maize. -
@narain and @lh3
I tried exactly the same stuff as described by you for SNPs and InDels detection and landed into same conclusion that Samtools misses a lot of SNPs and InDels otherwise pointed by IGV. Here are the details of my query: http://seqanswers.com/forums/showthread.php?t=26496 . I guess the answer is simple, there got to be better tools to extract SNPs and InDels from BAM files. Have you tried GATK / Dindel ?
AbiLeave a comment:
-
@lh3
Thank you for pointing out that this discussion has nothing to do with Bowtie2 vs BWA and I should address it in another thread. I noticed there is already a thread running which raises concern for using mpileup output via bcftools to get SNPs and InDels, and so I just raised my concern there itself. Here is the link: http://seqanswers.com/forums/showthr...034#post105034 . Do let me know what 'awk' command to use from the mpileup output if bcftools is not to be used. I have also sent an email to samtools-help mailing list as per your suggestion.Leave a comment:
-
I would suggest you make a new thread. You can also ask on the samtools-help mailing list.Leave a comment:
-
I'm glad to see that people stepped away from using terms like sensitivity, specificity, false positive rate, true positive rate, etc. All of those metrics rely on knowledge of true negative and false negative counts. So unless the simulation intentionally includes data that should NOT align then the expected negative count is always zero and therefore you can have no true negatives. Since TN is in the numerator of specificity then it will also always be 0. Since false positive rate is calculated as (fp)/(fp+tn) and true positive rate is (tp)/(tp+fn) it seems to me impossible to use those terms for these evaluations either.
It seems to be that precision and recall are more appropriate since they only rely on one's definition of "relevant returns". For a mapper one could define that as alignment placed within N bp of actual where N is whatever you think appropriate. Then the precision becomes (relevant_aligned)/(aligned_reads) (which is the same as (TP)/(TP+FP) AKA the positive predictive value) and recall is (properly aligned)/total_simulated_reads. Finally you can conveniently calculate the f-measure to combine these two values into a single value which is handy for making plots. I also like the look of plotting precision vs recall but I don't know if that's standard practice.Leave a comment:
-
Actually in the fermi paper, I used a table and three paragraphs to discuss indel calling, more than SNP calling. Increasingly more evidence suggests that for a high coverage sample, de novo assembly or local re-assembly based approaches usually perform better for indel calling.
Indels from the fermi alignment were called from raw mpileup output without any indel realignment models (for short reads, dindel, freebayes, samtools and gatk all use sophisticated indel realignment). I only used an awk one-liner to filter raw mpileup output. I use mummer and I know its dnadiff is simpler, but mummer does not work well with mammalian genomes and without SAM/BAM output, it is hard to visualize its alignment to investigate potential problems.
Readjoiner is very impressive. It was indeed faster than SGA. However, at least at the time of publication, it only implements graph generation. It lacks error correction, graph cleaning and scaffolding, which are less interesting theoretically but more important to good assembly in practice. These steps may also take more time and memory than graph generation. In addition, with a reimplementation of the BCR algorithm, both sga and fermi are much faster now than before. With multiple threads, they may take less wall-clock time than readjoiner for graph generation (readjoiner uses 51 hours to assemble 40X simulated human data according to its paper; fermi can build index and generate graph in 24 wall-clock hours for 36X real human data). Someone has also found a potentially faster prefix-suffix matching algorithm than readjoiner in terms of CPU time. As a note, fermi and SGA essentially index all the substrings of reads. In theory, they should be slower than readjoiner which only looks for prefix-suffix matches.
At last, I did not develope fermi as a pure de novo assembler. A few others (Jared, Richard and Anthony Cox) and I are more interested in exploring FM-index for other applications.Last edited by lh3; 05-13-2013, 11:12 AM.Leave a comment:
-
Someone told me that MUMmer has a hard-coded length limit something around 512Mbp, but I have not checked myself. Anyway, MUMmer uses about 14 bytes per base. Even if it does not have the length limit, it will need 40GB RAM to index human. That is why few use MUMmer to align against mammalian genomes (I think this is a well accepted conclusion). BWA and others use much less RAM for large genomes.
Because of the large RAM requirements, I have not tried MUMmer on short reads, but I doubt its post-processor nucmer is well tuned for short query sequences. Lacking the SAM output also adds works in comparisons. Nonetheless, in theory one can use MUMmer for seed alignment and post-process the maximal-exact matches reported by MUMmer to get an accuracy similar to other mappers and to generate SAM output. That requires a lot of additional works.
Even if you add SNPs and short indels to E. coli, aligning a genome to itself is too simple to evaluate the performance of a whole-genome aligner. Any reasonable genome aligners can trivially give the right result. There will be differences in the placement of indels, but that again is determined by how close the aligner's scoring matrix is to the simulator's matrix, but not by the true accuracy of aligners.Leave a comment:
-
BWA-MEM vs Nucmer
@lh3
I now read the paper that you pointed out to me earlier comparing different aligners:

It looks like BWA-MEM is the best choice for paired-end reads, and the best choice for longer reads. I notice in the paper that you mentioned that BWA-MEM scales well to large genomes than Nucmer. What do you really mean by this as you have not supported this by any data or figure ?
Also, you compared Nucmer with BWA-MEM only for genome size data of 4.6 Mb. Would it also make sense to include Nucmer in short read aligner comparisons , and see how it performs ?
Further, to check the accuracy of Nucmer and BWA-MEM for large size data such as 4.6 Mb, would it not be more meaningful to align a genome to itself and see which one of the two tools give lesser substitution ? This might be better than aligning two different strain genome to check accuracy. Essentially this is the same approach you adopted for comparing different short-read aligners using simulated reads of human genome to align to human genome itself.
NarainLeave a comment:
-
EDIT: I will not add new post to this thread. Just explain more clearly why we should not compare CIGARs in case others fall into the same pitfall. Firstly, we all know that in a tandem repeat, multiple CIGARs are equivalent. It is not clear from the text whether shi et al have considered this. Secondly, CIGAR is greatly affected by the scoring system used in simulation and in alignment. Two examples:
CGATCAGTAGCA......GCTAG-ATACGCAC
CGATCA--TGCA......GCTAGCATA-GCAC
versus
CGATCAGTAGCA......GCTAGAATCGCAC
CGATCA-T-GCA......GCTAGCAATGCAC
Which alignment is "optimal" is solely determined by mismatch, gap open and gap extension penalties. If the simulator simulates gaps under an edit-distance model, a mapper with a more realistic affine-gap penalty model will look worse. Comparing CIGARs on simulated data is essentially comparing which mapper uses a scoring system closer to the simulator. It has little to do with the true alignment accuracy. Thirdly, most simulation assumes the entire read can be aligned, but in reality, real data can be clipped. CIGAR comparison bias towards glocal alignment, but forcing glocal alignment may lead to false variant calls. Introducing clipping to ~1% of reads has little effect on downstream analyses. Finally, the rate of different CIGARs is several orders of magnitude higher than the mapping error rate. When we compare CIGARs, we are ignoring mapping errors, the errors we care about most. To this end, the ROC curves (Figure 4) in shi et al. have measured neither mapping nor alignment accuracy. The figure does not prove subread is more accurate for genomic read mapping. More proper evaluation of mapping accuracy shows that subread produces errors 100 times more than the competitors at a similar sensitivity.
It makes absolute sense to me to compare the accuracy of aligners in terms of CIGAR correctness in addition to the correct mapping location. That's certainly not a pitfall. In this way, the accuracy of aligners can be measured in the highest resolution. I can not see how could this cause a problem for dealing with the tandem repeat sequences.
It does not make sense to me that your scoring system affects the values of a CIGAR string. The CIGAR just tells you matches/mismatches, insertions, deletions, unmapped bases, irrelevant to your scoring system.
It is an irresponsible way to say that Subread is 100 times less accurate than your aligner without having concrete and convincing evidence. It is absolutely fine for me that you do not like our aligners. But the discussion should be conducted in a professional way that will help advance this field.Leave a comment:
-
For the purpose of comparison, I also attach our evaluation results. Clearly, remarkable differences can be observed between the results from different evaluation studies. Briefly, our evaluation had the following features:
(1) A read had to have a correct CIGAR string in addition to having the correct mapping location if was called a correctly mapped reads (all aligners had relatively low accuracy with this stringent criteria).
(2) Real quality scores (from a SEQC data) was used in our simulator (read bases were mutated according to their quality scores), giving rise to a simulation dataset highly similar to the real Illumina sequencing data.
(3) Read length is 100bp.
(4) SNPs and indels were introduced to the reference genome before simulated reads were extracted.
(5) A third-party simulation software (Mason) was also included in our evaluation.
For more information about our evaluation, please refer to http://www.ncbi.nlm.nih.gov/pubmed/23558742
 Last edited by shi; 04-26-2013, 08:17 PM.
Last edited by shi; 04-26-2013, 08:17 PM.Leave a comment:
-
It seems that this discussion is going nowhere. I will leave the ROC curves for subread along with other mappers given single-end data. Let's users and readers decide which mappers to use. Command lines can be found here. Some mappers are slower than I would expect probably because the average sequencing error rate is high. If any mapper is misused, please let me know. Thank you. PS: If you are concerned with uniform error rate and my biased view, you can also find other ROC curves from the Bowite2 paper and the LAST paper [PMID:23413433].
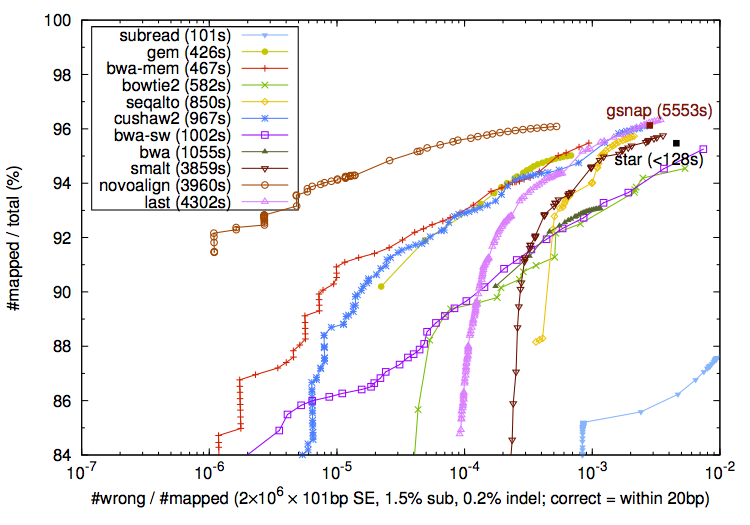
EDIT: I will not add new post to this thread. Just explain more clearly why we should not compare CIGARs in case others fall into the same pitfall. Firstly, we all know that in a tandem repeat, multiple CIGARs are equivalent. It is not clear from the text whether shi et al have considered this. Secondly, CIGAR is greatly affected by the scoring system used in simulation and in alignment. Two examples:
CGATCAGTAGCA......GCTAG-ATACGCAC
CGATCA--TGCA......GCTAGCATA-GCAC
versus
CGATCAGTAGCA......GCTAGAATCGCAC
CGATCA-T-GCA......GCTAGCAATGCAC
Which alignment is "optimal" is solely determined by mismatch, gap open and gap extension penalties. If the simulator simulates gaps under an edit-distance model, a mapper with a more realistic affine-gap penalty model will look worse. Comparing CIGARs on simulated data is essentially comparing which mapper uses a scoring system closer to the simulator. It has little to do with the true alignment accuracy. Thirdly, most simulation assumes the entire read can be aligned, but in reality, real data can be clipped. CIGAR comparison bias towards glocal alignment, but forcing glocal alignment may lead to false variant calls. Introducing clipping to ~1% of reads has little effect on downstream analyses. Finally, the rate of different CIGARs is several orders of magnitude higher than the mapping error rate. When we compare CIGARs, we are ignoring mapping errors, the errors we care about most. To this end, the ROC curves (Figure 4) in shi et al. have measured neither mapping nor alignment accuracy. The figure does not prove subread is more accurate for genomic read mapping. More proper evaluation of mapping accuracy shows that subread produces errors 100 times more than the competitors at a similar sensitivity. Furthermore, simulating realistic quality or not has little to do with the difference between my ROC and shi et al's ROC curves. Bowtie2 and LAST both simulate realistic quality and their ROC curves show 100-fold lower mapping error rate than shi's version.
EDIT2: Ah, have seen shi's latest reply. I am lost... Shi, in the other simulator thread, do you have any idea about why SNPs tend to be more clustered in a coalescent process? In this thread, do you know multiple CIGARs can be equivalent depending on where you put the gaps in a tandem repeat? Do you understand CIGARs are alignments and all alignments are determined by scoring, either explicitly or implicitly? Have you read my two alignment examples? Can you say confidently which is correct? I was assuming you knew at least some of these as a mapper developer. Furthermore, what makes you think my plot is not concrete or convincing? Because it uses uniform error rate? Then how would you explain why my plot is not much different from the ones in the LAST paper where the authors use realistic quality? Have you seen other 8 mappers in my plot? Why they are all doing well except subread? Isn't this a problem? If I masked mapper names, would you think the lightblue curve has any chance to match others on realistic simulation? Am I supposed to bias towards other mappers only to bush subread? Have you thought about why I only take on subread while openly sing praise for bowtie 2 (since beta 5; the discussions at the beginning of this thread is mostly about beta2 or so), gsnap, gem and novoalign? Why haven't I criticized the bowtie2, LAST and GEM papers when they all show bwa-backtrack is inferior? No one knows everything. Just learn. Finally, it is good for me to know I am unprofessional on sequence alignment. (Um.. probably I am unprofessional on engaging this discussion. Sorry.)Last edited by lh3; 04-27-2013, 05:35 AM.Leave a comment:
-
Originally posted by zee View Post
Thanks for your comments, zee.
The reason why Subread mapped more RNA-seq reads is because it has the ability to map exon-spanning reads. The seed-and-vote paradigm employed by Subread allows to determine the mapping location of the read using any part of the read sequence and Subread chooses the location mapped by the largest mappable region in the read. We estimated that in a typical RNA-seq dataset there is around 20 percent of reads that are exon-spanning reads and that is roughly the percentage difference shown in Figure 2.
So the extra mapped reads by Subread were not incorrect alignments, but those exon-spanning reads which were successfully Subread. This was further demonstrated by the comparison results shown in Tables 6 and 7. In this comparison, Subjunc was compared to TopHat 2, TopHat and MapSplice using both simulation data and the SEQC data. Subjunc uses the same mapping paradigm as used by Subread, but it performs complete alignment for exon-spanning reads and also outputs discovered exon-exon junctions. The read mapping locations reported by Subread are virtually the same as those reported by Subjunc, but Subjunc gives the full CIGAR information for junction reads but Subread only gives you the CIGAR for the largest mappable region in each junction reads (soft clipping for unmapped regions). Tables 6 and 7 show that Subjunc achieved excellent accuracy in exon-exon junction detection and in the mapping of RNA-seq reads, therefore this supported the high accuracy shown elsewhere for Subread in the paper.
WeiLeave a comment:
-
I think the mapping accuracy for RNA-seq data is equally important for that for gDNA-seq data. If you got wrong RNA-seq mapping results, how could you get a reliable downstream analysis done to get differentially expressed genes? And how could you accurately call SNPs (important for allele-specific expression analysis), indels and junctions? In some sense, finding genomic mutations from RNA-seq data is more important than doing that for gDNA-seq data because those mutations are likely to be functional.Originally posted by lh3 View PostLeave a comment:
-
As I have told you before, the difference in mapping error rate in Subread study vs Bowtie2 study was mainly because we checked the correctness of the CIGAR strings. The known truth included in the simulation data allows you to perform such a precise comparison, which I think is really important for assessing the accuracy of aligners in the highest resolution. It is also important to check if aligners can correctly map the end bases because wrong SNP calls often come from those bases.Originally posted by lh3 View Post
I do not agree with your statement "When there is an indel at the last couple of bases of a read, there is no way to find the correct CIGAR". Subread uses all the indels it found in the dataset to perform a realignment for reads which have indels at its end. This is in principle similar to what GATK does.
I also do not agree with your statement "The mainstream indel calling pipelines, including samtools, gatk and pindel, all use realignment. They do not trust CIGAR." Why do you need to generate CIGAR strings at the first place if you do not trust them? I think the the realignments performed by gatk etc are based on the CIGAR strings provided by the aligners. So they do trust the CIGAR, but they try to do a better job.
I do not understand "In addition, if we worry about CIGAR, we can do a global alignment to reconstruct it, which is very fast." If you can do this very fast, why dont you do it at the first place?Leave a comment:
-
The complexity of cancer is clearly demonstrated in the diverse ecosystem of the tumor microenvironment (TME). The TME is made up of numerous cell types and its development begins with the changes that happen during oncogenesis. “Genomic mutations, copy number changes, epigenetic alterations, and alternative gene expression occur to varying degrees within the affected tumor cells,” explained Andrea O’Hara, Ph.D., Strategic Technical Specialist at Azenta. “As...07-08-2024, 03:19 PM
Leave a comment: