 Tweet
Tweet
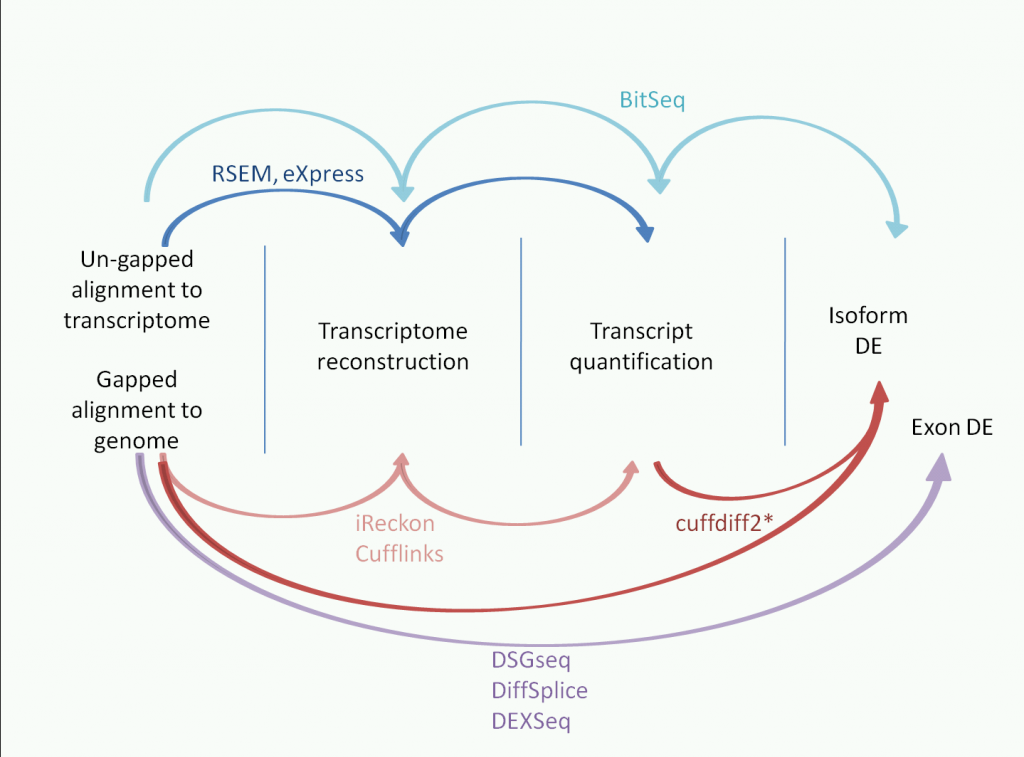
iReckon summary:
iReckon generates isoforms and quantifies them. However, this is based on gapped alignment to the genome (unlike eXpress, RSEM and BitSeq which are based on alignments to the transcriptome). It doesn't have a built in DE function, so each sample is run separately.
This tool is a little curious because it requires both a gapped alignment to the genome, and the unaligned reads in fastq or fasta format with a reference genome. Since it requires a BWA executable, it's doing some re-alignment. iReckon claims to generate novel isoforms with low false positives by taking into consideration a whole slew of biological and technical biases.
One irritating thing in getting the program running is that you need to re-format your refgene annotation file using an esoteric indexing tool from the Savant genome browser package. If you happen to use IGV, this is a bit tedious. Apparently, this will change in the next version. Also, iReckon takes up an enormous amount of memory and scratch space. For a library with 350 million reads, you would need about 800 G scratch space. Apparently everything (run time, RAM, and space) is linear to the number of reads, so this program would be a alright for a subset of the library or for lower coverage libraries.
iReckon generates isoforms and quantifies them. However, this is based on gapped alignment to the genome (unlike eXpress, RSEM and BitSeq which are based on alignments to the transcriptome). It doesn't have a built in DE function, so each sample is run separately.
This tool is a little curious because it requires both a gapped alignment to the genome, and the unaligned reads in fastq or fasta format with a reference genome. Since it requires a BWA executable, it's doing some re-alignment. iReckon claims to generate novel isoforms with low false positives by taking into consideration a whole slew of biological and technical biases.
One irritating thing in getting the program running is that you need to re-format your refgene annotation file using an esoteric indexing tool from the Savant genome browser package. If you happen to use IGV, this is a bit tedious. Apparently, this will change in the next version. Also, iReckon takes up an enormous amount of memory and scratch space. For a library with 350 million reads, you would need about 800 G scratch space. Apparently everything (run time, RAM, and space) is linear to the number of reads, so this program would be a alright for a subset of the library or for lower coverage libraries.

Comment