 Tweet
Tweet
I was in need for a FastQ editor that is fast, has a GUI and works also on Windows. Also PREFERABLY without dependencies like Java/DotNet. After some quite long search I couldn't find one so I decided to build my own. I am a biologist but I have some programming skills also.
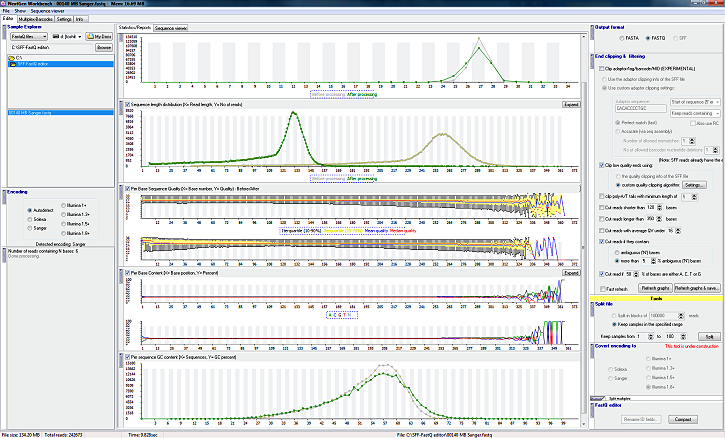
Until now I have a full SFF/FastQ viewer with some editing options also:
* Detect low quality (untrusted) ends
* Clip untrusted ends
* Trim poly-A/T tails with minimum length of x bases
* Cut reads shorter than x bases
* Cut reads longer than x bases
* Cut reads with average QV under specified threshold
* Cut reads if they contain N bases (I can specify how many)
* Cut read if x% of bases are either A, C, T or G
* Split multiplexed files (barcode detection via perfect match or assembly)
* Read lister - Show all reads (no matter how many they are). It can show: Read name, Base sequence, average quality, sequence length
* Graphic quality representation for each individual read. Color coded.
* Sequence length distribution graph
* Per base sequence quality graph
* Per base GC content graph
* Per base sequence content graph
* Per base N content graph (integrated in the 'Per Base Content' graph)
* Per sequence quality scores graph graph
The memory footprint for opening any file (no matter how large) should never exceed 30MB.
The program is free of course.
Feedback will be highly appreciated.
Until now I have a full SFF/FastQ viewer with some editing options also:
* Detect low quality (untrusted) ends
* Clip untrusted ends
* Trim poly-A/T tails with minimum length of x bases
* Cut reads shorter than x bases
* Cut reads longer than x bases
* Cut reads with average QV under specified threshold
* Cut reads if they contain N bases (I can specify how many)
* Cut read if x% of bases are either A, C, T or G
* Split multiplexed files (barcode detection via perfect match or assembly)
* Read lister - Show all reads (no matter how many they are). It can show: Read name, Base sequence, average quality, sequence length
* Graphic quality representation for each individual read. Color coded.
* Sequence length distribution graph
* Per base sequence quality graph
* Per base GC content graph
* Per base sequence content graph
* Per base N content graph (integrated in the 'Per Base Content' graph)
* Per sequence quality scores graph graph
The memory footprint for opening any file (no matter how large) should never exceed 30MB.
The program is free of course.
Feedback will be highly appreciated.

Comment