 Tweet
Tweet
Hello Members,
I need your guidance in selecting k-mer for assembling bacterial genome, paired end using SPAdes.
I know and have read section
SPAdes is smart enough to select its own K-mer, and assemble thereafter.
My question is:
I'm automating assembly and downstream analysis for isolates over 1000. I'd like to understand how to select an optimal K-mer based on read length.
If I were to set K-mer based on read length, what should be those? And why?
Currently, I'm having Illumina 150X2 data.
K-mer(s) may differ based on coverage too. But lets say I've data of coverage less than 30X.
I came across below URL too.
Also, there are tools like k-mer genie and velvet_optimizer. (I've not tried them yet)
There's no recipe for these (k-mer, coverage) kind of situations, but there has to be an optimal way which might help to have reasonable output and results?
I need your guidance in selecting k-mer for assembling bacterial genome, paired end using SPAdes.
I know and have read section

SPAdes is smart enough to select its own K-mer, and assemble thereafter.
My question is:
I'm automating assembly and downstream analysis for isolates over 1000. I'd like to understand how to select an optimal K-mer based on read length.
If I were to set K-mer based on read length, what should be those? And why?
Currently, I'm having Illumina 150X2 data.
K-mer(s) may differ based on coverage too. But lets say I've data of coverage less than 30X.
I came across below URL too.
Also, there are tools like k-mer genie and velvet_optimizer. (I've not tried them yet)
There's no recipe for these (k-mer, coverage) kind of situations, but there has to be an optimal way which might help to have reasonable output and results?

 Other than that there are not really any strict rules, just the longer the read, and the more coverage, generally the longer the kmer you can use. SPAdes, as far as I know, does not select a kmer length, but rather makes a combined assembly (by default) using multiple pre-selected kmer lengths of 55, 33, and 21. I think these values are low and a higher value, particularly for the max, would be better, at least for 150bp reads and good coverage.
Other than that there are not really any strict rules, just the longer the read, and the more coverage, generally the longer the kmer you can use. SPAdes, as far as I know, does not select a kmer length, but rather makes a combined assembly (by default) using multiple pre-selected kmer lengths of 55, 33, and 21. I think these values are low and a higher value, particularly for the max, would be better, at least for 150bp reads and good coverage.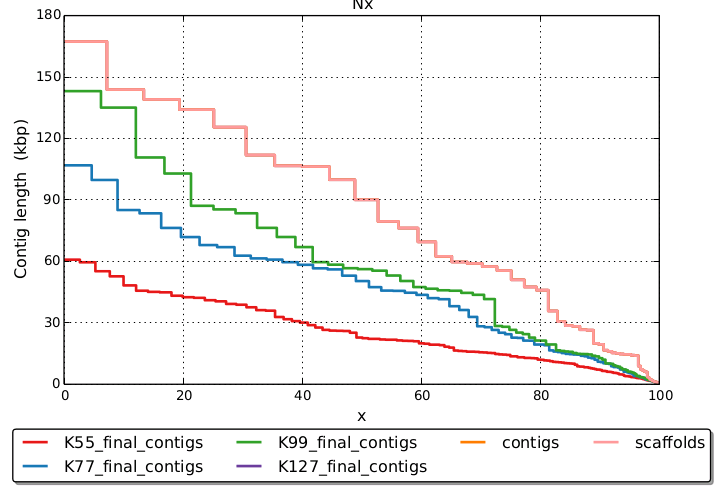
Comment