-
It depends of course on what you're trying to assemble. I've had good luck with microbial BAC clones assembling completely just with fragment (paired-end) data- those are only ~100-150kb. Microbial genomes will probably give you a bunch of contigs.Originally posted by bio_informatics View Post
Here's an example of fragment data only for 2.3Mb microbial genome, showing bigger contigs with increasing Kmer:
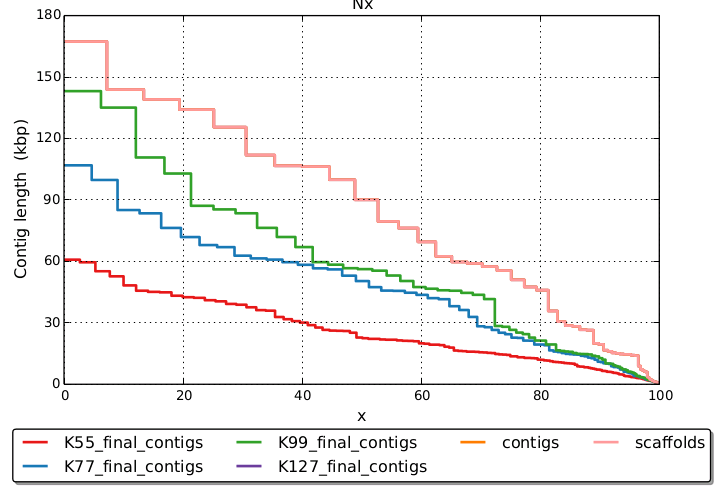
(final 'scaffolds' overlays final 'contigs' because there's no scaffolding without mate pair)
cheers- ScottLast edited by milw; 04-15-2015, 05:16 AM.Leave a comment:
-
Hi Brian,
Thanks for your valuable points. Definitely, k-mer won't be the read length, (un)fortunately :P
Definitely, k-mer won't be the read length, (un)fortunately :P
That's correct, SPAdes makes a combined assembly based on k-mer used.
But then again, the k-mer used are governed by the read length. Hence, was my question.
I wanted to understand - should I let SPAdes predict its usage of k-mer which it identifies by read length. OR, should I check read length and based on it, I can run as (example from its documentation):
As suggested by milw, and chipper; I should be attempting mentioned practices.spades.py -k 21,33,55,77 -
Thanks.Leave a comment:
-
Hi Chipper,
Oh, yes; I was oblivious to this feature. Thank you for reminding.
Originally posted by Chipper View PostLeave a comment:
-
Hi Milw,
Thank you for sharing your experience.
I do not have mate paired data. I hope that should not make a huge difference for k-mer outputs and resulting assembly?
I'll definitely try with 99 kmer and 127 as you've.
Originally posted by milw View PostLast edited by bio_informatics; 04-14-2015, 10:02 AM.Leave a comment:
-
I've been doing a lot of bacterial assemblies with SPades 3.5, and in all cases I've seen using kmer 99 or 127 works best in terms of contig # and N50. This has been with 2x 150 fragment PE plus a mate pair library. I had one trial set of K12 data that had fewer misassemblies using K77 than with K99 or 127.Leave a comment:
-
You can restart SPAdes with the addition of new k-mers if you think the assembly is not good enough. It combines the results from multiple k-mers, to me that seems like the best approach.Leave a comment:
-
The optimal kmer-length should be less than read length Other than that there are not really any strict rules, just the longer the read, and the more coverage, generally the longer the kmer you can use. SPAdes, as far as I know, does not select a kmer length, but rather makes a combined assembly (by default) using multiple pre-selected kmer lengths of 55, 33, and 21. I think these values are low and a higher value, particularly for the max, would be better, at least for 150bp reads and good coverage.
We have not had a good experience with tools that automatically try to determine the best kmer length based on kmer frequency histograms, and I don't think there is any theoretical validity to that approach. Running multiple assemblies with different kmer lengths, and selecting the one with the best metrics, seems like the best approach - at least, if you are using a fast assembler, like Velvet. SPAdes is too slow for that approach.Leave a comment:
-
SPAdes: selecting K-mer based on read length
Hello Members,
I need your guidance in selecting k-mer for assembling bacterial genome, paired end using SPAdes.
I know and have read section
SPAdes is smart enough to select its own K-mer, and assemble thereafter.
My question is:
I'm automating assembly and downstream analysis for isolates over 1000. I'd like to understand how to select an optimal K-mer based on read length.
If I were to set K-mer based on read length, what should be those? And why?
Currently, I'm having Illumina 150X2 data.
K-mer(s) may differ based on coverage too. But lets say I've data of coverage less than 30X.
I came across below URL too.
Also, there are tools like k-mer genie and velvet_optimizer. (I've not tried them yet)
There's no recipe for these (k-mer, coverage) kind of situations, but there has to be an optimal way which might help to have reasonable output and results?Last edited by bio_informatics; 04-14-2015, 05:08 AM.

-
The complexity of cancer is clearly demonstrated in the diverse ecosystem of the tumor microenvironment (TME). The TME is made up of numerous cell types and its development begins with the changes that happen during oncogenesis. “Genomic mutations, copy number changes, epigenetic alterations, and alternative gene expression occur to varying degrees within the affected tumor cells,” explained Andrea O’Hara, Ph.D., Strategic Technical Specialist at Azenta. “As...07-08-2024, 03:19 PM
Leave a comment: