 Tweet
Tweet
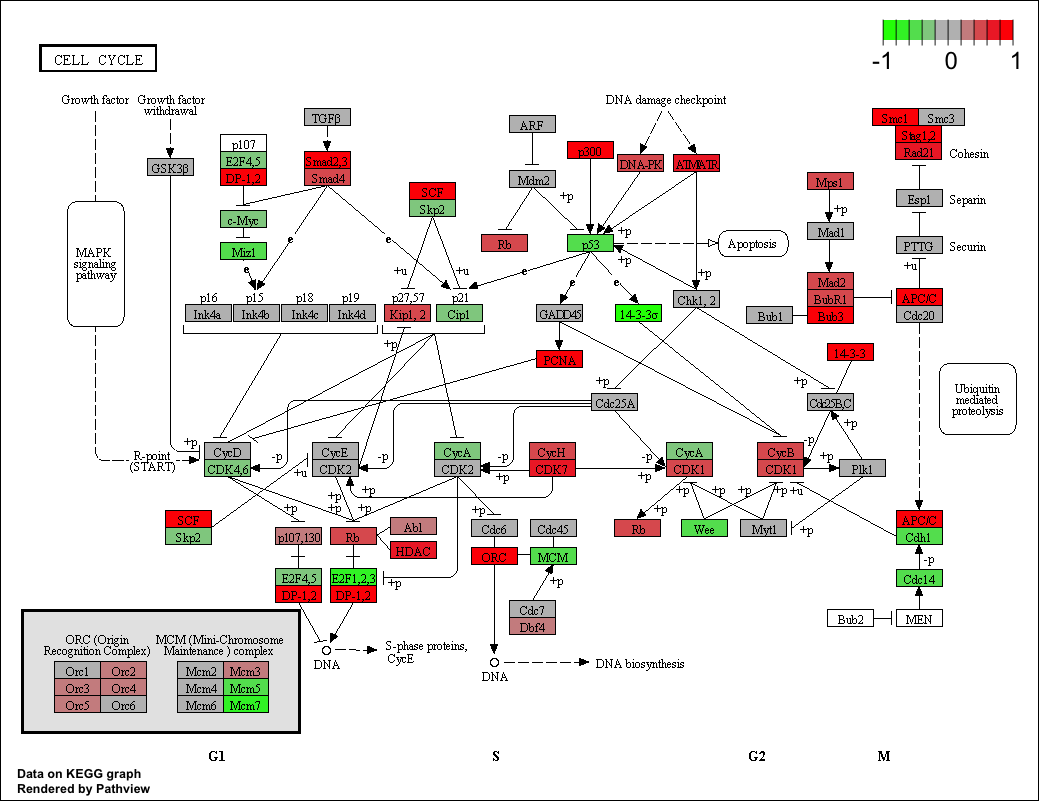
The gage package (2.12.0) now includes a new tutorial, "RNA-Seq Data Pathway and Gene-set Analysis Workflows". Note you need to update to current release versions of R(3.0.2)/ Bioconductor(2.13) to use all the features. Please check it out:
We first cover a full workflow from preparation, reads counting, data preprocessing, gene set test, to pathway visualization in about 40 lines of codes. The same workflow can be used for GO analysis or other types of gene set analysis too. We also describe joint workflows, i.e. to do gene-level analysis using one of the major RNA-Seq analysis tools, DEseq/DEseq2, edgeR, limma and Cufflinks, and feed the results into GAGE/Pahview for pathway analysis or visualization. All these workflows are implemented in R/Bioconductor.
Comments and questions are welcome. Thanks!

We first cover a full workflow from preparation, reads counting, data preprocessing, gene set test, to pathway visualization in about 40 lines of codes. The same workflow can be used for GO analysis or other types of gene set analysis too. We also describe joint workflows, i.e. to do gene-level analysis using one of the major RNA-Seq analysis tools, DEseq/DEseq2, edgeR, limma and Cufflinks, and feed the results into GAGE/Pahview for pathway analysis or visualization. All these workflows are implemented in R/Bioconductor.
Comments and questions are welcome. Thanks!
Comment