 Tweet
Tweet
Hello,
I tried to get gene expression level bby using tophat/cufflinks pipeline. I follow the step exactly like the flowchart in this link:
I have no problem until step cuffquant and I get an error when I trried to do the cuffnorm step. The error said something about reconstituted expression bundle does not match GTF. I don't understand why this is happen. If there are anyboddy who has experience maybe you can share with me about this problem. And also, I'm quite confused about the GTF reference paraameter in each step. Which one we must used from UCSC/ensemble, which one we used from the transcript.gtf as a result of cufflinks, and which one we must use the merged gtf from cuffmerge. I will really appreciate for your experience sharing if you would because I'm really new to this tool.
For me,
cufflinks -> I use GTF from UCSC
cuffmerge -> I still use GTF from UCSC
cuffquant -> I use merged GTF from cuffmerge
cuffnorm -> I use GTF from cuffmerge
Thank you all.
I tried to get gene expression level bby using tophat/cufflinks pipeline. I follow the step exactly like the flowchart in this link:
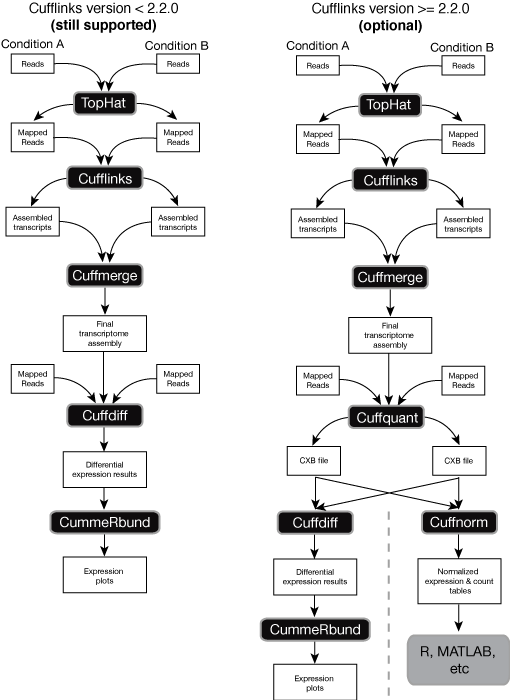
I have no problem until step cuffquant and I get an error when I trried to do the cuffnorm step. The error said something about reconstituted expression bundle does not match GTF. I don't understand why this is happen. If there are anyboddy who has experience maybe you can share with me about this problem. And also, I'm quite confused about the GTF reference paraameter in each step. Which one we must used from UCSC/ensemble, which one we used from the transcript.gtf as a result of cufflinks, and which one we must use the merged gtf from cuffmerge. I will really appreciate for your experience sharing if you would because I'm really new to this tool.
For me,
cufflinks -> I use GTF from UCSC
cuffmerge -> I still use GTF from UCSC
cuffquant -> I use merged GTF from cuffmerge
cuffnorm -> I use GTF from cuffmerge
Thank you all.