
Originally posted by yasutake
View Post
reference :
>chr1
gaagcgtagtctagtcagcgtagtcgtaactcat
MAQ format:
I326_2_FC306FCAAXX:8:18:1354:1553 chr1 597 - 0 0 99 99 99 0 01 0 35 GGTGGGACCGTTCGTGAAGGCTGGCCCATTGAGGA GGBFPOLQOPLHYRDOCLYM`OO^VSP``YR`_]T
I326_2_FC306FCAAXX:8:22:173:821 chr1 597 - 0 0 99 99 99 0 0 10 35 GGTGGGACCGTTCGTGAAGGCTGGCCCATTGAGGA NHELOUPVTZUT_WU^```````````````````
I326_2_FC306FCAAXX:8:29:309:1409 chr1 597 - 0 0 99 99 99 0 01 0 35 GGTGGGACCGTTCGTGAAGGCTGGCCCATTGAGGA HLHOBQCOOEGTP]LNJXVX`````J`````````
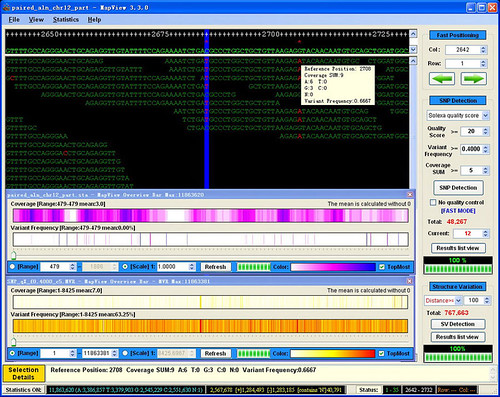
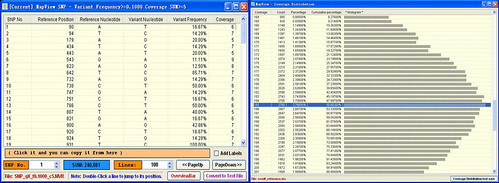
you can input these 2 files to make a mvf format file in mapview.
confirm that there is only one reference id , if there are multiple reference id ,please first use splitter to split the reference and alignment file










Leave a comment: