 Tweet
Tweet
Hi all,
I am trying to analyse my PE Illumina data using tophat.
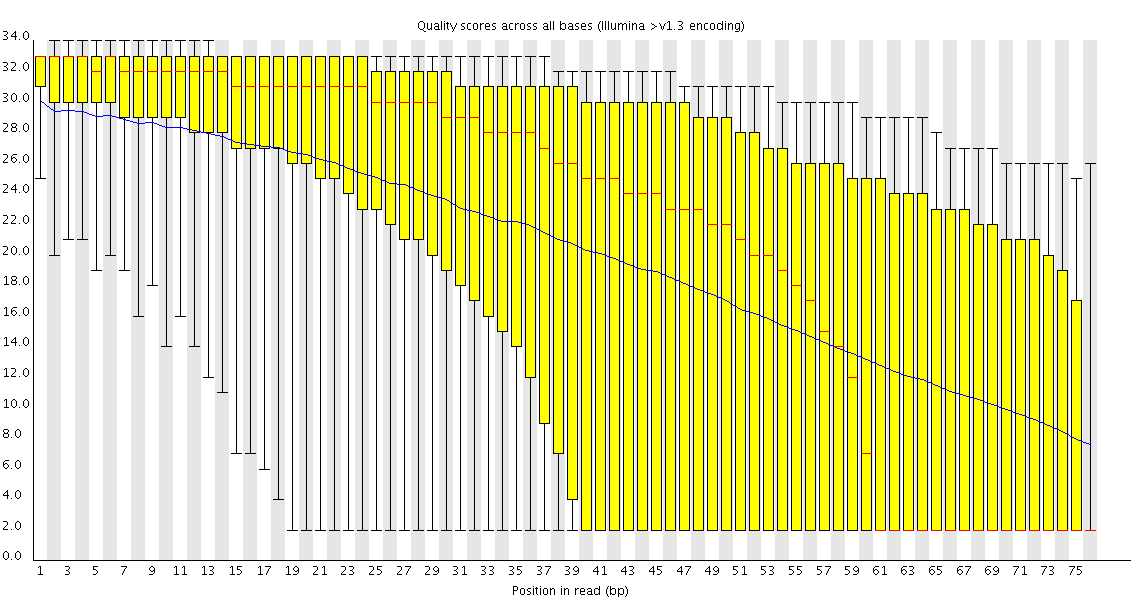
At first I run fastqc. Checking the raw data, I discovered at the beginnings (and presumably at the ends) of my reads I have some containments from the adapters of the sequencing.
I run bowtie first on both the full length and trimmed sequences and got better results with the trimmed sequences.
Do I need to trim the data before running tophat?
Does someone know how to do it? do I need to convert my trimmed sam files (bowtie output) back into fastq files?
Thanks for any help
Assa
I am trying to analyse my PE Illumina data using tophat.
At first I run fastqc. Checking the raw data, I discovered at the beginnings (and presumably at the ends) of my reads I have some containments from the adapters of the sequencing.
I run bowtie first on both the full length and trimmed sequences and got better results with the trimmed sequences.
Do I need to trim the data before running tophat?
Does someone know how to do it? do I need to convert my trimmed sam files (bowtie output) back into fastq files?
Thanks for any help
Assa
Comment